


How the FDA Regulates Medical AI
And a case study on how to successfully build it!

Machine learning is starting to play a bigger role in diagnosing and treating patients: well over 100 ML-based medical devices have been approved by the US’s Food and Drug Administration (FDA) in the past 5 years.
Thus far, the FDA has dealt with regulating these ML-based medical devices in a fairly ad hoc manner. However, now that it’s clear that ML in healthcare is here to stay, the FDA has started putting together a formal framework on how they are going to regulate ML-based medical devices going forward. They released a proposed regulatory framework for ML-based medical devices and an action plan for improvements and are now trying to finalize their regulatory approach.
For anyone building ML-based medical devices, it’s extremely important to understand the FDA’s proposed regulatory framework to ensure that you are building your product in a compliant manner. In addition, since the regulatory framework hasn’t been finalized yet, this is also a great opportunity to give feedback to the FDA and ensure that the right regulations get put into place.
However, in order to understand the FDA’s proposed regulatory framework, you need to first understand in-depth how to build software and hardware-based medical devices in regulated environments. The goal of this article is to provide this necessary context: we’ll use a case study to walk through the high-level process you’ll need to follow to build an FDA-regulated, ML-based medical device, presupposing no knowledge about the FDA.
What is Different About Regulating Machine Learning?
Before we jump in, it’s worth understanding what makes regulating machine learning so different from what the FDA has traditionally regulated: drugs and hardware-based medical devices.
Generally speaking, it’s easier for regulators if the things they’re regulating make large, infrequent changes rather than many small, frequent changes. This is because, traditionally, each modification to the drug or medical device has required an entire new application. Thus, large and infrequent changes minimize the amount of paperwork for both the FDA and the company creating the technology.
Drugs are easy to work with for this reason because they take a long time to develop and rarely have minor modifications. It normally takes around ten years for a drug to get to market, and once it’s in the market, the only real changes that happen are significant overhauls once every decade or so. Because the “cycle time” of drugs is so long, the FDA has plenty of time to approve them. The FDA implements a fairly straightforward premarket review of drugs: whenever a new drug or a modified drug comes onto the market, carefully assess whether it is safe and effective. This premarket approval process can take around 10 months.
Hardware-based medical devices are in a similar boat: minor modifications are infrequent, since you can’t easily change hardware once it’s launched. Thus, the FDA’s high level framework of reviewing every new and modified hardware-based medical device works just fine.
But this framework starts to break down when we consider software-based medical devices, called SaMDs. These are devices like chronic disease management apps, like Livongo and Onduo, or digital therapeutics, like Akili’s EndeavorRx. These software solutions can be updated practically every day, whenever someone writes a new line of code. Some of these modifications may be significant, like adding new functionality to the device, while others may not, like changing the color of a button. Which of these modifications are consequential enough to require FDA review?
This question becomes even more challenging with machine learning-based SaMDs. These are devices like Viz.ai’s platform that uses AI to detect signs of a stroke in CT scans and alert on-call stroke specialists. These ML-based SaMDs can update literally every time new data comes in, and the ML model itself might be a black box.
Taking months to review every change to an ML-based SaMD is impossible, since by the time the review has finished, the ML model will have already changed thousands of times. And yet, you can’t not regulate changes to ML-based SaMDs; you have to ensure that they remain safe and effective after every modification. So what’s the way out?
The FDA has been grappling with this question for years, and it’s the reason that regulating ML-based SaMDs has been such a difficult task.
What is Software as a Medical Device?
We’ve talked about SaMDs, but before we go any further, it’s helpful to clear up some potential confusion that I’ve seen around the term.
The term Software as a Medical Device (SaMD) describes software that’s supposed to be used for medical purposes — such as diagnosing, monitoring, mitigating, or curing diseases or other conditions — without needing specialty hardware. A good example is Nines' NinesAI system, which helps radiologists by triaging CT scans that show signs of a stroke or a brain tumor. It counts as a SaMD since it uses software (of course) to help diagnose patients, and it runs on commodity hardware like laptops.
However, software that’s intended to drive a hardware medical device (e.g. software that controls a pacemaker) is not a SaMD, since it’s the hardware, not the software, that’s driving medical outcomes. This class of software is instead called Software in Medical Devices (SiMDs).
Similarly, software that automates administrative work in a healthcare setting is also not considered a SaMD, since it’s not being directly used for a medical purpose. What’s more, the FDA doesn’t currently regulate software that helps with general fitness, health, or wellness, since that software is considered low risk.
Process for Building ML-Based SaMDs
Let’s build out an example case study to walk through the high-level process of building an ML-based SaMD.
Let’s say that we are developing a mobile app — call it DermoDx — that helps dermatologists determine whether a skin lesion is benign or malignant. This app takes pictures of the lesion using the smartphone’s camera, runs the images through a ML model to determine the lesion’s physical characteristics, and provides this information to a dermatologist. DermoDx counts as a SaMD because it’s software that’s used for medical purposes (namely, helping to diagnose skin cancer) and runs on commodity hardware (namely, a phone).
So how do we successfully develop the app, get FDA signoff, and deploy it?
Step 1. Determine Risk Classes
First, we need to determine which risk classes our app falls into. These risk classes determine which type of form we need to submit, how much documentation we need to show the FDA, and more.
There are three main risk classification frameworks that are pertinent to ML-based SaMDs. These risk frameworks are relatively independent from each other: just because one risk framework categorizes the SaMD as low risk doesn’t mean that the others will as well.
Medical Device Risk Classes
Our first risk framework helps us figure out which application we need to submit to the FDA. It’s a generic framework that splits all medical devices into three risk classes: Class I devices pose the lowest risks to the patient, while Class III devices pose the highest risks.
Note that, no matter which risk class a device is in, the manufacturer still has to comply with the FDA’s general controls, which are rules around how to brand the device, maintain sanitary conditions in the device's manufacturing plant, and more.
Class I (low risk):
Class I devices pose low risk to the patient. Some examples of Class I medical devices include floss, bandages, and software to retrospectively analyze glucose monitor data. Today, about 47% of medical devices fall under this category, and 95% of these are exempt from the FDA’s regulatory process.
For these Class I devices, we will have to submit one of the following applications:
No application: No application is needed if our device is exempt from the regulatory process; we simply have to register our device with the FDA.
510k: We’ll submit a 510k application if there’s already a device similar to ours on the market. This is a short application that details our device’s performance and how it compares to a similar device that is already on the market.
De Novo: We’ll submit a De Novo application if we have a novel device (novel means we are the first device of this kind on the market). This is a longer application with a lot more performance testing.
Any device that’s been registered with the FDA, including one exempt from regulation, is called FDA registered. A device that’s accepted through the 510k or De Novo pathway is called FDA cleared.
I’ve summarized this information into this table:

Class II (medium risk):
Class II devices pose medium risk to the patient. Some examples of Class II medical devices include pregnancy test kits, the Apple Watch ECG app, and most clinical decision support software. About 43% of medical devices currently fall under this category.
In addition to general controls, Class II devices have to comply with special controls, which are additional rules that differ based on the type of device you are building. Similar to Class I devices, Class II devices require either no application, a 510k, or a De Novo. The main difference is that very few Class II devices can get by with no application.
Our app, DermoDx, would likely fall into Class II, since it’s providing vital information to the dermatologist for a patient in a serious condition — but the dermatologist is still making the ultimate diagnosis. Let’s assume that there’s already an app that helps with skin cancer diagnosis on the market; in this case, we’ll submit a 510k.

Class III (high risk):
Class III devices pose a high risk to the patient. Some examples of Class III medical devices include pacemakers, heart valves, and software that makes treatment decisions for patients in critical conditions (this is what my research shows, but I’m waiting on the FDA to confirm this). About 10% of medical devices currently fall in this category.
For these Class III devices, we will have to submit one of the following applications:
510k: We’ll submit a 510k if a device similar to ours is already on the market.
Premarket Approval (PMA): We’ll submit a PMA application if we have a novel device; this is a very long application, with tons of performance testing and many more controls.

Software Risk Classes
Now that we’ve figured out which application we need to submit, we need to figure out how much documentation we need to provide. We figure this out by identifying which software risk class our app, DermoDx, falls into. Class A contains the software with the lowest risk to patients, while Class C contains software with the highest risk. Again, this system is independent of the previous class system: this one is only about medical software, while the previous one covers all medical devices.
Class A (low risk):
A piece of medical software (whether SaMD or SiMD) falls in Class A if the software can’t lead to any injuries if the software malfunctions, or we can put in risk control measures outside of the software to prevent it from accidentally injuring anyone. For example, software that stores a patient’s historical blood pressure information for a provider to review is Class A software since, even if the software incorrectly stores the patient’s past blood pressure levels, the provider can just be told to recheck the patient’s current blood pressure before taking any action.
For Class A software, the following documentation is required:

Class B (medium risk):
A software system falls in Class B if the software can lead to non-serious injuries even with external risk control measures in place. For example, software that controls syringes would be considered Class B software, since, if the software malfunctions, the provider might accidentally draw a bit too much blood, but the provider can easily pull the syringe out of the patient and avoid causing any serious injuries.
For Class B software, the following documentation is required:

Class C (high risk):
A software system falls in Class C if the software can lead to serious injuries or death even with external risk control measures in place. Software that makes any diagnosis or treatment decisions for patients in serious conditions is usually considered Class C, since any mistakes by the software can seriously hurt the patient.
For Class C software, the following documentation is required:

Our app, DermoDx, would likely fall into Class C, since a misdiagnosis from the dermatologist based on wrong information from our app could cause us to fail to diagnose skin cancer — which could have serious or even fatal consequences — or treat the patient for a disease that they don’t actually have.
SaMD Risk Categories
The third class system we use is the FDA’s SaMD risk categorization framework, which categorizes our app based on its intended use. The FDA guidance is still a bit weak on the exact purpose of this framework; I asked them for clarification but haven’t heard back yet.
Anyway, the two major factors that determine the intended use of a SaMD are:
How significant the information provided by the SaMD is to the healthcare decision: whether information is used to treat or diagnose, to drive clinical management, or just to inform clinical management.
The state of the healthcare situation or condition: whether the patient is in a critical, serious, or non-serious situation or condition (e.g. whether you’re diagnosing cancer or a splinter).
We use these factors to place our app into one of four categories, from lowest (I) to highest risk (IV). The higher the significance and the more critical the condition, the higher the risk. (Note, again, that these categories are totally independent of the Class I/II/III and Class A/B/C systems we talked about before.)

Some examples of SaMDs in different categories are:
Category I (lowest risk): A SaMD stores historical blood pressure information for a provider to review. This SaMD is in Category I because it’s just aggregating data and won’t trigger any immediate or near-term actions for the patient.
Category II (medium-low risk): A SaMD predicts risk scores for patients for developing stroke or heart disease. It’s in Category II because it’s used to detect early signs of a disease, but treating the condition isn’t time-critical.
Category III (medium-high risk): A SaMD uses the microphone of a smart device to detect interrupted breathing during sleep and wakes up the patient. This SaMD is in Category III because it’s used to treat the patient, though the patient isn’t in a time-critical condition.
Category IV (highest risk): A SaMD analyzes CT scans to make treatment decisions in patients with acute stroke. It’s in Category IV because the patient is in a critical condition and time is of the essence.
Our app, DermoDx, will likely “drive clinical management” in a “serious healthcare situation” since it’s aiding the dermatologist in identifying malignant skin lesions, which is a potentially fatal disease, though our intervention isn’t as time-sensitive as say, treating someone experiencing a stroke or a heart attack. Thus, our app would be placed in Category II.
In summary, we have now identified that DermoDx is a Class II, Class C, and Category II SaMD. This has helped us determine which application to submit, how much documentation we need to write, and more. Now we can now move on to actually building our app!
Step 2. Get Optional (But Highly Recommended!) Feedback
Submit Pre-Submission
Once our paperwork is in order, we can choose to get FDA feedback on our submission plans and ask questions through the pre-submission process. The pre-submission is completely optional, but it’s a great opportunity for us to get feedback on topics like which application we need to submit (510k vs. De Novo vs. PMA), whether our planned clinical studies are adequate, and whether we’re doing our statistical analyses properly. Plus, the pre-submission process is completely free!
Request Payer Feedback
We can also shorten the time between the FDA clearing or approving our device and a payer agreeing to reimburse it by starting conversations with payers now.
There are two opportunities for us to get input from payers:
Early Payer Feedback Program: We can get input from payers on our clinical trial design and other plans for gathering clinical evidence.
Parallel Review with CMS: We can get feedback from both the FDA and Center for Medicare and Medicaid Services (CMS) on our clinical trial design. They’ll concurrently and independently review the clinical trial results in our application.
Step 3. Develop Your ML-Based SaMD
Establish Quality System and Good Machine Learning Practices
Before we start developing our SaMD, we need to establish a quality system for our company. This is a group of processes, procedures, and responsibilities that’ll enable us to build a high quality product, which we will then document in our quality management system (QMS) repository. For example, our QMS might contain documents on the procedure we use to manufacture a device, our process for investigating and resolving complaints about our device, and details on different employee roles and responsibilities.
We also have to demonstrate that we are using the FDA’s “Good Machine Learning Practices,” which are just a set of ML best practices recommended by the agency. The FDA is still working with organizations around the world to develop these practices, but they’ve released a few so far:
Appropriately separate training, validation, and test datasets.
Use data that’s relevant to the clinical problem that you are trying to solve.
Provide users with an appropriate level of transparency about the output and algorithm.
Conduct Hazard Analysis
While developing the app, we need to brainstorm all of the hazards that our app might create. For each of these hazards, we assign a risk level based on the probability of the event occurring and the severity of the harm it causes. Then, we implement mitigations until the risk level is below a certain cut-off point that we’ve defined for ourselves.
For our app, DermoDx, let’s say that the app uses AI to identify the skin lesion, calculates the width of it, and displays the width on the app to help the dermatologist make a diagnosis. Thus, we might identify:
Hazard: Width of a skin lesion is incorrectly calculated and and thus incorrectly displayed
Sequence of events: Dermatologist sees the incorrect width of the skin lesion and jumps to an incorrect conclusion
Hazardous situation: Dermatologist misdiagnoses patient
Severity: Severe
Probability: Medium
Harm: Patient doesn’t get necessary treatment or gets treatment that’s not needed
To decrease the probability of this hazardous situation occurring, we might identify potential mitigations as:
Display a digital ruler next to the lesion so that the dermatologist can double-check the width of the lesion themselves.
Notify the dermatologist if the computed width is a statistical outlier and ask them to recheck by measuring the width of the patient’s skin lesion in person.
Run Verification and Validation Tests
While we are building our app, we need to write verification and validation test cases to ensure that we are building the app correctly, and we need to document the results in our quality management system.
Verification tests are quite like the unit tests and integration tests that are familiar to most software developers: they test every part of the code to verify that it works as intended. Validation tests, meanwhile, are more high-level and test whether the product meets the high-level product requirements. One way to conduct validation tests is to go to customers and have them use the product without guidance.
Clinically Evaluate Device
Once our app is built, we need to evaluate if it actually performs well. We do this by demonstrating:
Valid clinical association: Is there a valid clinical association in the research between our app’s results and our target clinical condition? For DermoDx, we can look at existing clinical research or do our own clinical trials to prove that looking at physical characteristics of the skin lesion is actually useful in identifying malignant skin lesions.
Analytical validation: Does our app work and produce accurate, reliable, and precise results? For DermoDx, we can use the evidence generated through our verification and validation tests to prove that the physical characteristics of the lesions that our app computes are accurate.
Clinical validation: Is our app producing clinically useful information? For DermoDx, we could share sensitivity and specificity percentages to show that the physical characteristics we are outputting are useful to a dermatologist trying to decide whether or not a skin lesion is malignant. To give some insight into the difference between analytical and clinical validation, our app could accurately measure the length of my thumb and thus pass the analytical validation tests. However, it would fail the clinical validation tests since the length of my thumb has no bearing on whether or not my skin lesion is malignant.
Step 4. Submit Application to the FDA
Submission Pathways
Now that we built and clinically evaluated our SaMD, we are ready to submit an application to the FDA! As you may recall, the application we need to submit (if any) depends on the risk classes of our device, whether or not our device is exempt, and whether a device similar to ours is already on the market. To summarize our earlier rules, we’ll have to submit one of the following:
No application: No application is needed if our device is exempt from the regulatory process; we simply have to register our device with the FDA.
510k: We’ll submit a 510k application if there’s already a device similar to ours on the market. This is a short application that details our device’s performance and how it compares to a similar device that is already on the market.
De Novo: We’ll submit a De Novo application if we have a novel device (novel means we are the first device of this kind on the market). This is a longer application with a lot more performance testing.
Premarket Approval (PMA): We’ll submit a PMA application if we have a novel device; this is a very long application, with tons of performance testing and many more controls.
Since our app, DermoDx, is a Class II medical device and there are already similar devices on the market, we would have to submit a 510k.
Predetermined Change Control Plan
As we discussed at the beginning of this piece, under traditional rules, we’d have to go back to the FDA for re-review every time we change our SaMD. And since our SaMD uses ML, we’d have to be going back to the FDA nonstop.
The way to avoid constantly asking the FDA for review modifications is to submit a Predetermined Change Control Plan (PCCP). In this plan, we specify:
SaMD Pre-Specifications (SPS): How we plan on modifying our device in the future.
Algorithm Change Protocol (ACP): What procedures we’re going to follow when making these changes.
Then, once our device is on the market, any changes that lie within the bounds of our change control plan don’t need to go to the FDA for approval. We simply have to document that we made the change.
Obviously, not all types of modifications to the SaMD can be put into our change control plan. The ML model automatically updating whenever it gets new data is fine for the PCCP, but modifying our product to start diagnosing melanoma is far outside the bounds. Unfortunately, the line between changes that are acceptable within the PCCP and those that aren’t is still quite blurry at the moment, so we’d probably want to ask the FDA for more clarity.
Going back to our app, DermoDx, we could propose two potential modifications in our change control plan:
Increasing the app’s ML model’s sensitivity and specificity by using real-world data
Extending the app to also work on more platforms, such as adding Android if we start out only supporting iPhones.
For each of these proposed modifications, we would include details about how we’d collect and store data, retrain our ML models, evaluate the performance of the model, communicate changes to our users, and more. Here’s a potential outline for our document:
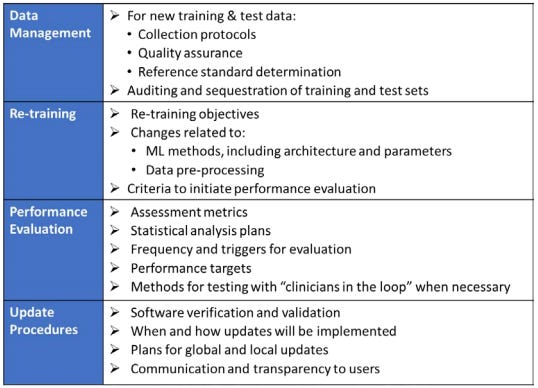
Step 5. Deploy ML-Based SaMD
Device Modifications
Finally, we’ve built our app, our application has been accepted by the FDA, and the app is on the market! So how do we modify the app?
Depending on the type of modification, we’ll have to either just document the change, edit the PCCP, or submit a brand-new application for FDA review (i.e. redo Step 4).
Unfortunately, the FDA’s guidance on when to use each approach is still quite weak. Generally, minor changes that don’t change the functionality of the device or introduce new risks, such as adding additional encryption or access control, can just be documented. The FDA has released some guidance that explains what changes count as minor enough for documentation for 510ks and De Novos as well as for PMAs but the lines are still a little blurry.
If our change is not minor enough to just be documented, we’ll have to check whether the modification we are making is within the bounds of any SPS and ACPs in our PCCP. If it is, then we can simply document our proposed modification.
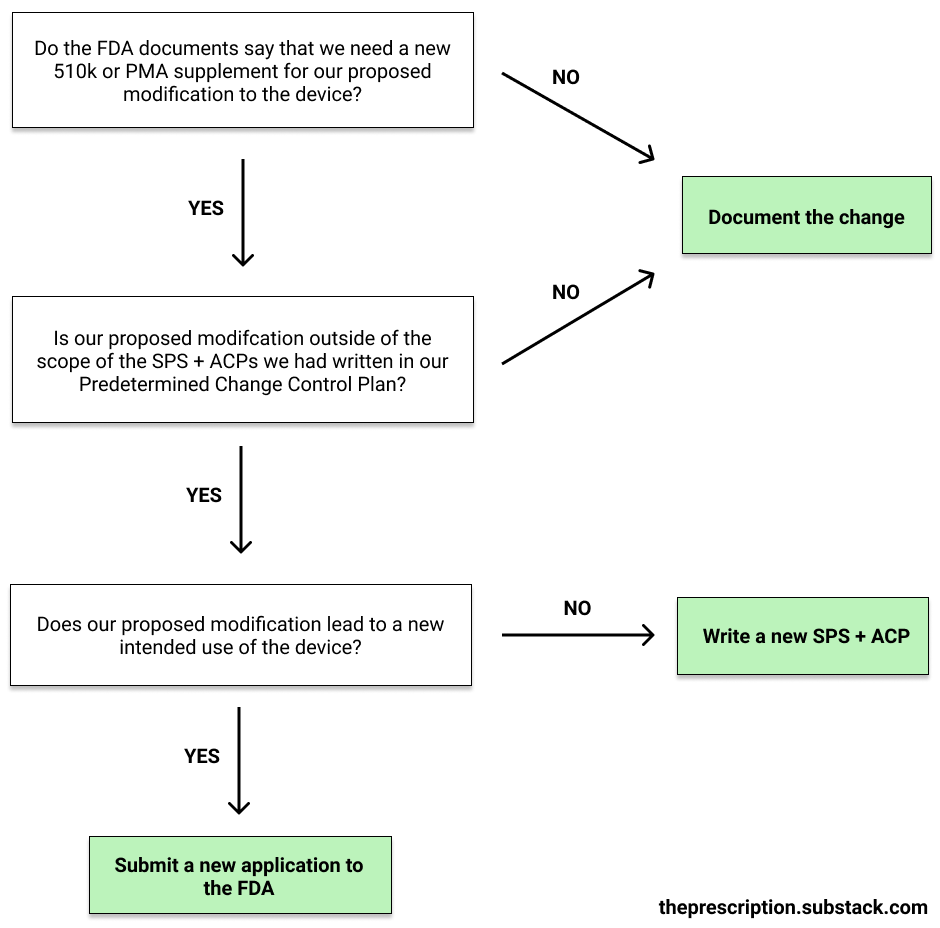
If the change wasn’t in our PCCP, we’ll check if our modification changes the intended use of our device. We do this by looking at the SaMD Risk Categories (covered in Step 1) and seeing if our device has now moved risk categories. If it hasn’t, we can just write a new SPS and ACP to cover the change that we’re planning to make. If it has moved categories, our modification is beyond the intended use that our ML-based SaMD was previously authorized for, and thus we have to submit a whole new application to the FDA.
For DermoDx, Let’s say there are a couple of types of modifications we want to make.
Scenario 1: Increase sensitivity and specificity of the app’s ML model, consistent with PCCP
Based on the real-world data we collected, we want to improve the performance of our ML model. We follow all the steps we laid out in the control change plan to retrain the model with this additional data and ensure that the new model performs well. Since we’re within the scope of the change control plan we laid out, we can make this modification without additional FDA review.
Scenario 2: Extend app to work with Android phones, consistent with PCCP
Suppose our app started out as iPhone-only, but now we want it to work on Android too. Again, we follow all the steps we laid out on the change control plan to ensure the app works as intended on Android. Since we are within the scope of the plan we laid out, we can make this modification without additional FDA review.
Scenario 3: Make the app patient-facing, inconsistent with PCCP
Currently, only dermatologists are using our app to aid them in identifying malignant skin lesions. Let’s say we want to extend our app to have a patient-facing portion. Since this modification completely changes our app’s intended use cases and introduces many new risks, we’ll need to submit a new application for this new version for FDA review.
Real World Monitoring
Our work isn’t done once our app is out in the real world: we’ll also need to monitor its performance on an ongoing basis. We’ll likely need to periodically report modifications we’ve made to the app and the app’s performance to the FDA.
Conclusion
This entire regulatory framework is still in flux, and the FDA is still figuring out their final approach towards regulating ML-based SaMDs. There’s still a lot that needs to be figured out — such as how to ensure that machine learning models work effectively in real world settings and are generalizable. The FDA is actively looking for feedback, so this is the best opportunity for us to contribute and ensure the right policies get put into place.
A huge thank you to Neel Mehta, Urvi Kanakaraj, Gabriel Straus, and Anand Sampat for answering all my questions and/or helping review this article!
Was this article helpful to you? Do you have any feedback or particular topics that you want me to cover? If so, I would love to hear it! Please comment below or email me at maitreyee.joshi@gmail.com!
🧠 you did such a good job of breaking down a tricky process.
An insightful read!